5.2 Summary metrics
Pierre Denelle, Boris Leroy and Maxime Lenormand
2026-07-21
Source:vignettes/a5_2_summary_metrics.Rmd
a5_2_summary_metrics.RmdIn this vignette, we describe two functions to compute summary metrics:
- metrics calculated for each species and/or site
site_species_metrics() - metrics calculated for each bioregion
bioregion_metrics()
1. Terminology clarification
The bioregion is focused on bioregionalization,
i.e. clustering of geographical areas on the basis of species data.
However, there are several cases where species can also become part of
the clustering (for example, in bipartite network clustering), which
poses terminology issues.
To be conceptually accurate, we have chosen to name species clusters as ‘chorotypes’:
Bioregion: A group of sites with similar species composition, identified through clustering analysis. Bioregions are geographic units.
Chorotype: A group of species with similar distributions within the study area. Chorotypes are biological units. This generally corresponds to the concept of “regional chorotype” sensu (Baroni Urbani et al., 1978), as clarified by (Fattorini, 2015). Note that when clustering on worldwide ranges, the concept becomes “global chorotypes” (see (Fattorini, 2015) for further details).
Possible cases of chorotypes
| Clustering scenario | Site clusters | Species clusters | Conceptual basis |
|---|---|---|---|
| Site-only clustering | Bioregions | — | Sites grouped by compositional similarity |
| Bipartite network clustering | Bioregions | Chorotypes (same cluster IDs) | Sites and species grouped by shared network structure |
| Species-only clustering | — | Chorotypes | Species grouped by distributional similarity |
| Post-hoc species assignment | Bioregions | Chorotypes (derived) | Species assigned to bioregions based on specificity/IndVal |
Bipartite network clustering
In bipartite network clustering, both sites and species are assigned to the same clusters (network modules). A species assigned to cluster 1 belongs to the same bioregion as sites assigned to cluster 1. We use the term chorotype to refer to the set of species assigned to a given bioregion, but it is important to understand that:
In bipartite clustering, bioregion ID = chorotype ID. They are two perspectives on the same network partition: bioregion refers to the sites in a cluster, chorotype refers to the species in that same cluster.
Site-only clustering with post-hoc species assignment
Species can be secondarily assigned to bioregions based on metrics such as maximum specificity or IndVal. Here, chorotype refers to the group of species most strongly associated with a given bioregion. Unlike bipartite clustering, this assignment is derived rather than intrinsic to the clustering algorithm.
2. Example data
We use the vegetation dataset included in the
bioregion.
data("vegedf")
data("vegemat")
# Calculation of (dis)similarity matrices
vegedissim <- dissimilarity(vegemat, metric = c("Simpson"))
vegesim <- dissimilarity_to_similarity(vegedissim)3. Bioregionalization
We use the same three bioregionalization algorithms as in the visualization
vignette, i.e., non-hierarchical, hierarchical, and network
bioregionalizations. In addition, we include a network
bioregionalization algorithm based on a bipartite network, which assigns
clusters to both sites and species. We chose three bioregions for the
non-hierarchical and hierarchical bioregionalizations.
# Non hierarchical bioregionalization
vege_nhclu <- nhclu_kmeans(vegedissim,
n_clust = 3,
index = "Simpson",
seed = 1)
vege_nhclu$cluster_info ## partition_name n_clust
## K_3 K_3 3
# Hierarchical bioregionalization
vege_hclu <- hclu_hierarclust(dissimilarity = vegedissim,
index = "Simpson",
seed = 1,
method = "average",
n_clust = 3,
optimal_tree_method = "best",
verbose = FALSE)
vege_hclu$cluster_info## partition_name n_clust requested_n_clust output_cut_height
## 1 K_3 3 3 0.5625
# Network bioregionalization
vege_netclu <- netclu_walktrap(vegesim,
index = "Simpson")
vege_netclu$cluster_info ## partition_name n_clust
## K_3 K_3 3
# Bipartite network bioregionalization
install_binaries(verbose = FALSE)
vege_netclubip <- netclu_infomap(vegedf,
seed = 1,
bipartite = TRUE)
vege_netclubip$cluster_info## partition_name n_clust
## K_8 K_8 84. Metric components
Before diving into specific metrics, we can understand the core terms using a simple example. Consider a study area with 4 sites and 4 species, where sites have been assigned to 2 bioregions.
4.1 Species-derived metrics
The following diagram shows the site-species matrix where sites are grouped by bioregion. Marginal sums give us all the core terms needed to compute metrics:
Species
sp1 sp2 sp3 sp4 n_b
┌─────┬─────┬─────┬─────┐
Site A │ 1 │ 1 │ · │ · │
B1 ───────┼─────┼─────┼─────┼─────┤ 2
Site B │ 1 │ 1 │ 1 │ · │
Bioregion ══════╪═════╪═════╪═════╪═════╪══════
Site C │ · │ 1 │ 1 │ 1 │
B2 ───────┼─────┼─────┼─────┼─────┤ 2
Site D │ · │ · │ 1 │ 1 │
└─────┴─────┴─────┴─────┘
n_sb sp1 sp2 sp3 sp4 n_b
(per bioregion) ┌─────┬─────┬─────┬─────┐
B1 │ 2 │ 2 │ 1 │ 0 │ 2
├─────┼─────┼─────┼─────┤
B2 │ 0 │ 1 │ 2 │ 2 │ 2
└─────┴─────┴─────┴─────┘
n_s (total) 2 3 3 2 n = 4
K_s (# bioreg) 1 2 2 1 K = 2| Term | Meaning | Where to find it |
|---|---|---|
| \(n\) | Total number of sites | Bottom-right corner (4) |
| \(K\) | Total number of bioregions | Bottom-right corner (2) |
| \(n_b\) | Sites in bioregion \(b\) | Right margin per bioregion row |
| \(n_s\) | Sites where species \(s\) occurs | Bottom margin per species column |
| \(K_s\) | Number of bioregions where species \(s\) occurs | Bottom margin \(n_s\) |
| \(n_{sb}\) | Sites in bioregion \(b\) with species \(s\) | The \(n_{sb}\) summary table |
Examples of calculations
From the \(n_{sb}\) table, all species-per-bioregion metrics follow directly:
Specificity (fraction of species’ occurrences in a bioregion): \[A_{sp1,B1} = \frac{n_{sp1,B1}}{n_{sp1}} = \frac{2}{2} = 1.00 \quad \text{(sp1 is exclusive to B1)}\] \[A_{sp2,B1} = \frac{n_{sp2,B1}}{n_{sp2}} = \frac{2}{3} = 0.67 \quad \text{(sp2 mostly in B1)}\]
Fidelity (fraction of bioregion’s sites with the species): \[B_{sp2,B1} = \frac{n_{sp2,B1}}{n_{B1}} = \frac{2}{2} = 1.00 \quad \text{(sp2 in all B1 sites)}\] \[B_{sp3,B1} = \frac{n_{sp3,B1}}{n_{B1}} = \frac{1}{2} = 0.50 \quad \text{(sp3 in half of B1)}\]
IndVal (indicator value = Specificity × Fidelity): \[IndVal_{sp1,B1} = 1.00 \times 1.00 = 1.00 \quad \text{(perfect indicator of B1)}\] \[IndVal_{sp2,B1} = 0.67 \times 1.00 = 0.67\]
4.2 Site-derived metrics
The following diagram shows the same site-species matrix, but now species are grouped by cluster (chorotype). We compute how many species from each cluster occur in each site:
Chorotypes
┌─── C1 ───┐ ┌─── C2 ───┐
sp1 sp2 sp3 sp4
┌─────┬─────┬─────┬─────┐
Site A │ 1 │ 1 │ · │ · │ 2
├─────┼─────┼─────┼─────┤
Sites Site B │ 1 │ 1 │ 1 │ · │ 3
├─────┼─────┼─────┼─────┤
Site C │ · │ 1 │ 1 │ 1 │ 3
├─────┼─────┼─────┼─────┤
Site D │ · │ · │ 1 │ 1 │ 2
└─────┴─────┴─────┴─────┘
n_c 2 2 n = 4
n_gc C1 C2 n_g
(per cluster) ┌───────┬───────┐
Site A │ 2 │ 0 │ 2
├───────┼───────┤
Site B │ 2 │ 1 │ 3
├───────┼───────┤
Site C │ 1 │ 2 │ 3
├───────┼───────┤
Site D │ 0 │ 2 │ 2
└───────┴───────┘
n_c 2 2 n = 4| Term | Meaning | Where to find it |
|---|---|---|
| \(n\) | Total number of species | Bottom-right corner (4) |
| \(n_c\) | Species in cluster \(c\) | Bottom margin per cluster |
| \(n_g\) | Species present in site \(g\) | Right margin per site row |
| \(n_{gc}\) | Species from cluster \(c\) present in site \(g\) | The \(n_{gc}\) summary table |
NOTE: in bipartite clustering, bioregion and chorotypes can be the exact same clusters. Nevertheless, we use different terms here to avoid confusion in the calculation of metrics.
Examples of calculations
Specificity of Site A for C1 (fraction of site’s species belonging to C1): \[A_{A,C1} = \frac{n_{A,C1}}{n_A} = \frac{2}{2} = 1.00 \quad \text{(Site A has only C1 species)}\]
Specificity of Site B for C1: \[A_{B,C1} = \frac{n_{B,C1}}{n_B} = \frac{2}{3} = 0.67 \quad \text{(Site B mostly has C1 species)}\]
Fidelity of Site A for C1 (fraction of C1 species present in Site A): \[B_{A,C1} = \frac{n_{A,C1}}{n_C1} = \frac{2}{2} = 1.00 \quad \text{(Site A has all C1 species)}\]
Fidelity of Site C for C1: \[B_{C,C1} = \frac{n_{C,C1}}{n_{C1}} = \frac{1}{2} = 0.50 \quad \text{(Site C has half of C1 species)}\]
5. List of site/species metrics included in the package
Metrics per cluster
When clusters are assigned to sites
(cluster_on = "site" or
cluster_on = "both")
| Metric | Entity | Cluster type | Based on | Occ | Ab | Formula (occurrence) | Interpretation |
|---|---|---|---|---|---|---|---|
| Specificity | Species | Bioregion | Co-occurrence | ✓ | ✓ | \(A_{sb} = \frac{n_{sb}}{n_s}\) | Fraction of species’ occurrences in bioregion |
| NSpecificity | Species | Bioregion | Co-occurrence | ✓ | ✓ | \(\bar{A}_{sb} = \frac{n_{sb}/n_b}{\sum_k n_{sk}/n_k}\) | Size-normalized specificity |
| Fidelity | Species | Bioregion | Co-occurrence | ✓ | ✓ | \(B_{sb} = \frac{n_{sb}}{n_b}\) | Fraction of bioregion’s sites with species |
| IndVal | Species | Bioregion | Co-occurrence | ✓ | ✓ | \(A_{sb} \times B_{sb}\) | Indicator value (specificity × fidelity) |
| NIndVal | Species | Bioregion | Co-occurrence | ✓ | ✓ | \(\bar{A}_{sb} \times B_{sb}\) | Size-normalized indicator value |
| Rho | Species | Bioregion | Co-occurrence | ✓ | ✓ | See section 7.1.1 | Standardized contribution index |
| CoreTerms | Species | Bioregion | Co-occurrence | ✓ | ✓ | \(n\), \(n_b\), \(n_s\), \(n_{sb}\) | Raw counts for custom calculations |
| Richness | Site | — | Co-occurrence | ✓ | — | \(S_g = n_g\) | Number of species |
| Rich_Endemics | Site | Bioregion | Co-occurrence | ✓ | — | \(E_g = \sum{K_s}\) | Number of endemic species in the site (i.e., species occurring in only one bioregion) |
| Prop_Endemics | Site | Bioregion | Co-occurrence | ✓ | — | \(\bar{PctEnd}_{g} = \frac{E_g}{S_g}\) | Proportion of endemic species in the site |
| MeanSim | Site | Bioregion | Similarity | — | — | \(\frac{1}{n_b - \delta} \sum_{g' \neq g} sim_{gg'}\) | Mean similarity to bioregion |
| SdSim | Site | Bioregion | Similarity | — | — | See section 7.2.1 | SD of similarity to bioregion |
When clusters are assigned to species
(cluster_on = "species" or
cluster_on = "both")
| Metric | Entity | Cluster type | Based on | Occ | Ab | Formula (occurrence) | Interpretation |
|---|---|---|---|---|---|---|---|
| Specificity | Site | Chorotype | Co-occurrence | ✓ | ✓ | \(A_{gc} = \frac{n_{gc}}{n_g}\) | Fraction of site’s species in cluster |
| NSpecificity | Site | Chorotype | Co-occurrence | ✓ | ✓ | \(\bar{A}_{gc} = \frac{n_{gc}/n_c}{\sum_k n_{gk}/n_k}\) | Size-normalized specificity |
| Fidelity | Site | Chorotype | Co-occurrence | ✓ | ✓ | \(B_{gc} = \frac{n_{gc}}{n_c}\) | Fraction of cluster’s species in site |
| IndVal | Site | Chorotype | Co-occurrence | ✓ | ✓ | \(A_{gc} \times B_{gc}\) | Indicator value (specificity × fidelity) |
| NIndVal | Site | Chorotype | Co-occurrence | ✓ | ✓ | \(\bar{A}_{gc} \times B_{gc}\) | Size-normalized indicator value |
| Rho | Site | Chorotype | Co-occurrence | ✓ | ✓ | See section 7.2.2 | Standardized contribution index |
| CoreTerms | Site | Chorotype | Co-occurrence | ✓ | ✓ | \(n\), \(n_c\), \(n_g\), \(n_{gc}\) | Raw counts for custom calculations |
Metrics in bioregionalization/clustering
These metrics summarize how an entity is distributed across all clusters, rather than in relation to each individual cluster.
When cluster_on = "site" (or "both")
| Metric | Entity | Based on | Occ | Ab | Formula | Interpretation |
|---|---|---|---|---|---|---|
| P | Species | Co-occurrence | ✓ | ✓ | \(1 - \sum_k \left(\frac{n_{sk}}{n_s}\right)^2\) | Evenness of species across bioregions (0–1) |
| Silhouette | Site | Similarity | — | — | \(\frac{a_g - b_g}{\max(a_g, b_g)}\) | Fit to assigned vs. nearest bioregion |
6. Usage
This section demonstrates how to use
site_species_metrics() with all metrics computed for both
sites and species. This is only possible in a bipartite network
clustering, where both sites and species receive clusters
simultaneously.
For this example, we will use the bipartite network bioregionalization from section 3, where both sites and species are assigned to the same clusters. We compute all available metrics for both sites and species.
all_metrics <- site_species_metrics(
bioregionalization = vege_netclubip,
bioregion_metrics = c("Specificity", "NSpecificity", "Fidelity",
"IndVal", "NIndVal", "Rho", "CoreTerms",
"Richness", "Rich_Endemics", "Prop_Endemics",
"MeanSim", "SdSim"), # You can also simply write "all"
bioregionalization_metrics = c("P", "Silhouette"),
data_type = "both",
cluster_on = "both",
comat = vegemat,
similarity = vegesim,
index = "Simpson",
verbose = FALSE)Typing the name of the object in the console calls
print(), which provides a concise overview of the output,
including the settings used, a preview of available metrics, and
instructions for accessing the data.
all_metrics## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: sites and species
## - Clustering data type: abundance
## - Metric data type: occurrence and abundance
##
## Computed metrics:
## - Per-cluster co-occurrence metrics (occurrence): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
## - Per-cluster co-occurrence metrics (abundance): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
## - Per-cluster richness & endemism metrics: Richness, Rich_Endemics, Prop_Endemics
## - Per-cluster similarity-based metrics: MeanSim, SdSim
## - Bioregionalization co-occurrence metrics (occurrence): P
## - Bioregionalization co-occurrence metrics (abundance): P
## - Bioregionalization similarity-based metrics: Silhouette
##
## Data preview:
## $species_bioregions (29576 rows x 20 cols):
## Species Bioregion n_sb n_s n_b Specificity_occ NSpecificity_occ Fidelity_occ
## 10017 1 392 551 515 0.711 0.148 0.761
## 10017 2 86 551 92 0.156 0.182 0.935
## 10017 3 68 551 75 0.123 0.176 0.907
## IndVal_occ NIndVal_occ Rho_occ w_sb w_s w_b Specificity_abund
## 0.542 0.113 -0.965 4545 10660 2831323 0.426
## 0.146 0.170 4.009 5349 10660 1136829 0.502
## 0.112 0.160 2.960 733 10660 137186 0.069
## NSpecificity_abund Fidelity_abund IndVal_abund NIndVal_abund Rho_abund
## 0.082 0.002 0.325 0.063 -9.237
## 0.542 0.005 0.469 0.507 15.718
## 0.091 0.005 0.062 0.083 -1.663
## # ... with 29573 more rows
##
## $species_bioregionalization (3697 rows x 3 cols):
## Species P_occ P_abund
## 10017 0.454 0.562
## 10024 0.463 0.504
## 10034 0.493 0.264
## # ... with 3694 more rows
##
## $site_chorotypes (5720 rows x 20 cols):
## Site Chorotypes n_gc n_g n_c Specificity_occ NSpecificity_occ Fidelity_occ
## 35 1 1 129 2219 0.008 0.003 0.000
## 35 2 121 129 873 0.938 0.892 0.139
## 35 3 7 129 430 0.054 0.105 0.016
## IndVal_occ NIndVal_occ Rho_occ w_gc w_g w_c Specificity_abund
## 0.000 0.000 -13.981 1 423 2873216 0.002
## 0.130 0.124 19.103 411 423 1149003 0.972
## 0.001 0.002 -2.237 11 423 103264 0.026
## NSpecificity_abund Fidelity_abund IndVal_abund NIndVal_abund Rho_abund
## 0.001 0 0.000 0.000 -7.973
## 0.948 0 0.135 0.131 11.313
## 0.051 0 0.000 0.001 -1.840
## # ... with 5717 more rows
##
## $site_chorological (715 rows x 3 cols):
## Site P_occ P_abund
## 35 0.117 0.055
## 36 0.465 0.199
## 37 0.350 0.082
## # ... with 712 more rows
##
## $site_bioregions (5720 rows x 7 cols):
## Site Bioregion Richness Rich_Endemics Prop_Endemics MeanSim SdSim
## 35 1 129 0 0.000 0.116 0.123
## 35 2 129 2 0.016 0.809 0.179
## 35 3 129 0 0.000 0.264 0.189
## # ... with 5717 more rows
##
## $site_bioregionalization (715 rows x 2 cols):
## Site Silhouette
## 35 0.540
## 36 0.379
## 37 0.511
## # ... with 712 more rows
##
## Access data with:
## your_object$species_bioregions
## your_object$species_bioregionalization
## your_object$site_chorotypes
## your_object$site_chorological
## your_object$site_bioregions
## your_object$site_bioregionalizationYou can also run summary() oçn the object to quickly see
a statistical summary for each output table, including the number of
rows and summary statistics for numeric columns.
summary(all_metrics)##
## Summary of site and species metrics
## ===================================
##
## Settings:
## - Number of partitions: 1
## - Cluster based on: sites and species
## - Clustering data type: abundance
## - Metric data type: occurrence and abundance
##
## Partition 1: Single partition (8 bioregions, 8 chorotypes)
## ----------------------------------------------------------
##
## Species-per-bioregion metrics ($species_bioregions):
## Metric Min Mean Median Max
## n_sb 0.000 0.000 0.0000000 500.000
## n_s 1.000 72.000 72.0000000 671.000
## n_b 1.000 15.000 15.0000000 515.000
## Specificity_occ 0.000 0.000 0.0000000 1.000
## NSpecificity_occ 0.000 0.000 0.0000000 1.000
## Fidelity_occ 0.000 0.000 0.0000000 1.000
## IndVal_occ 0.000 0.000 0.0000000 0.844
## NIndVal_occ 0.000 0.000 0.0000000 0.996
## Rho_occ -18.940 -0.335 -0.3346268 24.233
## w_sb 0.000 0.000 0.0000000 22716.000
## w_s 1.000 422.000 422.0000000 27353.000
## w_b 5558.000 55402.000 55402.0000000 2831323.000
## Specificity_abund 0.000 0.000 0.0000000 1.000
## NSpecificity_abund 0.000 0.000 0.0000000 1.000
## Fidelity_abund 0.000 0.000 0.0000000 0.161
## IndVal_abund 0.000 0.000 0.0000000 0.973
## NIndVal_abund 0.000 0.000 0.0000000 1.000
## Rho_abund -12.483 -0.239 -0.2393045 26.693
##
## Top species by IndVal_occ:
## 1. 12553 (Bioregion 2): 0.844
## 2. 12749 (Bioregion 1): 0.841
## 3. 11513 (Bioregion 1): 0.841
## 4. 11285 (Bioregion 2): 0.838
## 5. 13550 (Bioregion 2): 0.837
##
## Species summary metrics ($species_bioregionalization):
## Metric Min Mean Median Max
## P_occ 0 0.269 0.246 0.719
## P_abund 0 0.216 0.159 0.739
##
## Site-per-chorotype metrics ($site_chorotypes):
## Metric Min Mean Median Max
## n_gc 0.000 80.573 1.000 1091.000
## n_g 1.000 644.585 673.000 1221.000
## n_c 3.000 462.125 76.500 2219.000
## Specificity_occ 0.000 0.125 0.002 1.000
## NSpecificity_occ 0.000 0.125 0.028 1.000
## Fidelity_occ 0.000 0.091 0.015 1.000
## IndVal_occ 0.000 0.032 0.000 0.524
## NIndVal_occ 0.000 0.033 0.000 0.661
## Rho_occ -27.165 -0.927 -1.144 39.347
## w_gc 0.000 741.633 3.000 38926.000
## w_g 1.000 5933.064 4012.000 43753.000
## w_c 2942.000 530267.625 50655.500 2873216.000
## Specificity_abund 0.000 0.125 0.001 1.000
## NSpecificity_abund 0.000 0.125 0.010 1.000
## Fidelity_abund 0.000 0.001 0.000 0.496
## IndVal_abund 0.000 0.034 0.000 0.701
## NIndVal_abund 0.000 0.036 0.000 0.991
## Rho_abund -20.999 -0.462 -0.777 43.653
##
## Top sites by IndVal_occ:
## 1. 238 (Chorotype 2): 0.524
## 2. 433 (Chorotype 2): 0.522
## 3. 286 (Chorotype 2): 0.497
## 4. 86 (Chorotype 2): 0.488
## 5. 384 (Chorotype 2): 0.482
##
## Site chorological summary metrics ($site_chorological):
## Metric Min Mean Median Max
## P_occ 0 0.294 0.244 0.668
## P_abund 0 0.254 0.196 0.668
##
## Site-per-bioregion metrics ($site_bioregions):
## Metric Min Mean Median Max
## Richness 1 644.585 673.000 1221.000
## Rich_Endemics 0 1.185 0.000 71.000
## Prop_Endemics 0 0.002 0.000 0.156
## MeanSim 0 0.439 0.452 1.000
## SdSim 0 0.059 0.037 0.500
##
## Site summary metrics ($site_bioregionalization):
## Metric Min Mean Median Max
## Silhouette -1 -0.02 -0.029 0.789We can see it also displays the top sites or species for IndVal for a convenient quick look at our clustering structure.
You can also use str() to display the internal structure
of the object, showing the settings and the dimensions and column types
of each data frame component.
str(all_metrics)## bioregion.site.species.metrics object
## - Partitions: 1
## - Cluster based on: sites and species
## - Clustering data type: abundance
## - Metric data type: occurrence and abundance
## - Per-cluster co-occurrence metrics (occurrence): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
## - Per-cluster co-occurrence metrics (abundance): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
## - Per-cluster richness & endemism metrics: Richness, Rich_Endemics, Prop_Endemics
## - Per-cluster similarity-based metrics: MeanSim, SdSim
## - Bioregionalization co-occurrence metrics (occurrence): P
## - Bioregionalization co-occurrence metrics (abundance): P
## - Bioregionalization similarity-based metrics: Silhouette
##
## $ species_bioregions :'data.frame': 29576 obs. of 20 variables:
## ..$ Species : chr [1:29576] "10017" "10017" "10017" "10017" ...
## ..$ Bioregion : chr [1:29576] "1" "2" "3" "4" ...
## ..$ n_sb : num [1:29576] 392 86 68 1 2 1 0 1 153 74 ...
## ..$ n_s : num [1:29576] 551 551 551 551 551 551 551 551 232 232 ...
## ..$ n_b : int [1:29576] 515 92 75 26 4 1 1 1 515 92 ...
## ..$ Specificity_occ : num [1:29576] 0.71143 0.15608 0.12341 0.00181 0.00363 ...
## ..$ NSpecificity_occ : num [1:29576] 0.14806 0.18183 0.17636 0.00748 0.09726 ...
## ..$ Fidelity_occ : num [1:29576] 0.7612 0.9348 0.9067 0.0385 0.5 ...
## ..$ IndVal_occ : num [1:29576] 5.42e-01 1.46e-01 1.12e-01 6.98e-05 1.81e-03 ...
## ..$ NIndVal_occ : num [1:29576] 0.112695 0.169968 0.159897 0.000288 0.048628 ...
## ..$ Rho_occ : num [1:29576] -0.965 4.009 2.96 -9.04 -1.29 ...
## ..$ w_sb : int [1:29576] 4545 5349 733 1 2 18 0 12 3781 3549 ...
## ..$ w_s : int [1:29576] 10660 10660 10660 10660 10660 10660 10660 10660 7361 7361 ...
## ..$ w_b : int [1:29576] 2831323 1136829 137186 83829 26975 12303 8138 5558 2831323 1136829 ...
## ..$ Specificity_abund : num [1:29576] 4.26e-01 5.02e-01 6.88e-02 9.38e-05 1.88e-04 ...
## ..$ NSpecificity_abund: num [1:29576] 0.082265 0.541967 0.091103 0.000359 0.004661 ...
## ..$ Fidelity_abund : num [1:29576] 1.61e-03 4.71e-03 5.34e-03 1.19e-05 7.41e-05 ...
## ..$ IndVal_abund : num [1:29576] 3.25e-01 4.69e-01 6.23e-02 3.61e-06 9.38e-05 ...
## ..$ NIndVal_abund : num [1:29576] 6.26e-02 5.07e-01 8.26e-02 1.38e-05 2.33e-03 ...
## ..$ Rho_abund : num [1:29576] -9.24 15.72 -1.66 -2.73 -1.02 ...
## $ species_bioregionalization:'data.frame': 3697 obs. of 3 variables:
## ..$ Species: chr [1:3697] "10017" "10024" "10034" "10035" ...
## ..$ P_occ : num [1:3697] 0.4542 0.4631 0.4929 0.0588 0.3132 ...
## ..$ P_abund: num [1:3697] 0.5617 0.5037 0.26396 0.00308 0.05098 ...
## $ site_chorotypes :'data.frame': 5720 obs. of 20 variables:
## ..$ Site : chr [1:5720] "35" "35" "35" "35" ...
## ..$ Chorotypes : chr [1:5720] "1" "2" "3" "4" ...
## ..$ n_gc : num [1:5720] 1 121 7 0 0 0 0 0 241 585 ...
## ..$ n_g : num [1:5720] 129 129 129 129 129 129 129 129 867 867 ...
## ..$ n_c : int [1:5720] 2219 873 430 137 16 16 3 3 2219 873 ...
## ..$ Specificity_occ : num [1:5720] 0.00775 0.93798 0.05426 0 0 ...
## ..$ NSpecificity_occ : num [1:5720] 0.0029 0.8923 0.1048 0 0 ...
## ..$ Fidelity_occ : num [1:5720] 0.000451 0.138603 0.016279 0 0 ...
## ..$ IndVal_occ : num [1:5720] 3.49e-06 1.30e-01 8.83e-04 0.00 0.00 ...
## ..$ NIndVal_occ : num [1:5720] 1.31e-06 1.24e-01 1.71e-03 0.00 0.00 ...
## ..$ Rho_occ : num [1:5720] -13.981 19.103 -2.237 -2.268 -0.762 ...
## ..$ w_gc : int [1:5720] 1 411 11 0 0 0 0 0 4139 38926 ...
## ..$ w_g : int [1:5720] 423 423 423 423 423 423 423 423 43753 43753 ...
## ..$ w_c : int [1:5720] 2873216 1149003 103264 85285 16026 9049 3356 2942 2873216 1149003 ...
## ..$ Specificity_abund : num [1:5720] 0.00236 0.97163 0.026 0 0 ...
## ..$ NSpecificity_abund: num [1:5720] 0.000907 0.947603 0.05149 0 0 ...
## ..$ Fidelity_abund : num [1:5720] 3.48e-07 3.58e-04 1.07e-04 0.00 0.00 ...
## ..$ IndVal_abund : num [1:5720] 1.07e-06 1.35e-01 4.23e-04 0.00 0.00 ...
## ..$ NIndVal_abund : num [1:5720] 4.09e-07 1.31e-01 8.38e-04 0.00 0.00 ...
## ..$ Rho_abund : num [1:5720] -7.973 11.313 -1.84 -1.282 -0.431 ...
## $ site_chorological :'data.frame': 715 obs. of 3 variables:
## ..$ Site : chr [1:715] "35" "36" "37" "38" ...
## ..$ P_occ : num [1:715] 0.117 0.465 0.35 0.293 0.387 ...
## ..$ P_abund: num [1:715] 0.0553 0.1993 0.0825 0.0694 0.1883 ...
## $ site_bioregions :'data.frame': 5720 obs. of 7 variables:
## ..$ Site : chr [1:5720] "35" "35" "35" "35" ...
## ..$ Bioregion : chr [1:5720] "1" "2" "3" "4" ...
## ..$ Richness : num [1:5720] 129 129 129 129 129 129 129 129 867 867 ...
## ..$ Rich_Endemics: num [1:5720] 0 2 0 0 0 0 0 0 0 43 ...
## ..$ Prop_Endemics: num [1:5720] 0 0.0155 0 0 0 ...
## ..$ MeanSim : num [1:5720] 0.1156 0.8088 0.2635 0.0143 0.0291 ...
## ..$ SdSim : num [1:5720] 0.12299 0.17917 0.18876 0.01273 0.00742 ...
## $ site_bioregionalization :'data.frame': 715 obs. of 2 variables:
## ..$ Site : chr [1:715] "35" "36" "37" "38" ...
## ..$ Silhouette: num [1:715] 0.54 0.379 0.511 0.541 0.433 ...
## - attr(*, "n_partitions")= num 1
## - attr(*, "cluster_on")= chr "both"
## - attr(*, "clustering_data_type")= chr "abundance"
## - attr(*, "index_data_type")= chr "both"
## - attr(*, "has_similarity")= logi TRUE
## - attr(*, "has_comat")= logi TRUE
## - attr(*, "bioregion_metrics_occ")= chr [1:7] "Specificity" "NSpecificity" "Fidelity" "IndVal" ...
## - attr(*, "bioregion_metrics_abd")= chr [1:7] "Specificity" "NSpecificity" "Fidelity" "IndVal" ...
## - attr(*, "bioregionalization_metrics_occ")= chr "P"
## - attr(*, "bioregionalization_metrics_abd")= chr "P"
## - attr(*, "similarity_metrics")= chr [1:6] "Richness" "Rich_Endemics" "Prop_Endemics" "MeanSim" ...7. Metrics per cluster
7.1 Species-per-bioregion metrics
These metrics are computed when sites have clusters (i.e.,
cluster_on = "site" (or "both")). In the
following example, we compute all metrics
(bioregion_metrics = c("Specificity", "NSpecificity", "Fidelity", "IndVal", "NIndVal", "Rho", "CoreTerms")).
To compute these metrics, we need to provide comat.
7.1.1 Co-occurrence metrics: occurrence version
The occurrence metrics are computed when
data_type = "occurrence". By default, the function will
detect the type of data used for the clustering. However, this parameter
can be overriden by users, such that occurrence metrics can be
calculated for abundance clustering, and vice-versa. Users can also
specify data_type = "both" if they want to obtain both
versions of co-occurrence metrics.
nsb <- site_species_metrics(bioregionalization = vege_nhclu,
bioregion_metrics = c("Specificity", "NSpecificity",
"Fidelity", "IndVal", "NIndVal",
"Rho",
"CoreTerms"),
bioregionalization_metrics = NULL,
data_type = "occurrence",
cluster_on = "site",
comat = vegemat,
similarity = NULL,
index = NULL, # Name of similarity column
verbose = FALSE)
nsb## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: occurrence
## - Metric data type: occurrence
##
## Computed metrics:
## - Per-cluster co-occurrence metrics (occurrence): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
##
## Data preview:
## $species_bioregions (11091 rows x 11 cols):
## Species Bioregion n_sb n_s n_b Specificity_occ NSpecificity_occ Fidelity_occ
## 10001 1 27 254 358 0.106 0.056 0.075
## 10001 2 97 254 150 0.382 0.479 0.647
## 10001 3 130 254 207 0.512 0.465 0.628
## IndVal_occ NIndVal_occ Rho_occ
## 0.008 0.004 -15.645
## 0.247 0.310 8.384
## 0.321 0.292 9.722
## # ... with 11088 more rows
##
## Access data with:
## your_object$species_bioregionsSpecificity (occurrence)
The specificity \(A_{sb}\) of species \(s\) for bioregion \(b\) (De Cáceres & Legendre, 2009) is defined as
\[A_{sb} = \frac{n_{sb}}{n_s}\]
and measures the fraction of occurrences of species \(s\) that belong to bioregion \(b\). It therefore reflects the uniqueness of a species to a particular bioregion.
NSpecificity (occurrence)
A normalized version that accounts for the size of each bioregion is also available, as defined in (De Cáceres & Legendre, 2009):
\[\bar{A}_{sb} = \frac{n_{sb}/n_b}{\sum_{k=1}^K n_{sk}/n_k}\]
It corresponds to a normalized specificity value that adjusts for differences in bioregion size.
Fidelity (occurrence)
The fidelity \(B_{sb}\) of species \(s\) for bioregion \(b\) (De Cáceres & Legendre, 2009) is defined as
\[B_{sb} = \frac{n_{sb}}{n_b}\]
and measures the fraction of sites in bioregion \(b\) where species \(s\) is present. It therefore reflects the frequency of occurrence of a species within a bioregion.
IndVal (occurrence)
The indicator value \(IndVal_{sb}\) of species \(s\) for bioregion \(b\) can be defined as the product of specificity and fidelity (De Cáceres & Legendre, 2009):
\[IndVal_{sb} = A_{sb} \times B_{sb}\]
This index quantifies the strength of association between a species and a bioregion by combining its specificity (uniqueness to that bioregion) and fidelity (consistency of occurrence within that bioregion). High IndVal values identify species that are both frequent and restricted to a single bioregion, making them good indicators of that region.
NIndVal (occurrence)
A normalized version of the indicator value is also available:
\[\bar{IndVal}_{sb} = \bar{A}_{sb} \times B_{sb}\]
This normalization adjusts for differences in bioregion size, allowing more comparable indicator values across regions with unequal sampling effort or extent.
Rho (occurrence)
The contribution index \(\rho\) can also be calculated following (Lenormand et al., 2019):
\[\rho_{sb} = \frac{n_{sb} - n_s\frac{n_b}{n}}{\sqrt{\frac{n_b(n - n_b)}{n - 1} \frac{n_s}{n}(1 - \frac{n_s}{n}) }}\]
This index measures the deviation between the observed number of occurrences of species \(s\) in bioregion \(b\) and the expected value under random association, providing a standardized measure of contribution to the bioregional structure.
Co-occurrence metrics: abundance version
The occurrence metrics are computed when
data_type = "occurrence". By default, the function will
detect the type of data used for the clustering. However, this parameter
can be overriden by users, such that occurrence metrics can be
calculated for abundance clustering, and vice-versa.
The abundance version of these metrics can also be computed when
data_type = "abundance" (or
data_type = "both"). In this case the core terms and
associated metrics are:
- \(w_{sb}\) is the sum of abundances of species s in sites of bioregion b.
-
\(w_s\) is the total abundance of
species s.
- \(w_b\) is the total abundance of all species present in sites of bioregion b.
wsb <- site_species_metrics(bioregionalization = vege_nhclu,
bioregion_metrics = c("Specificity", "NSpecificity",
"Fidelity",
"IndVal", "NIndVal",
"Rho",
"CoreTerms"),
bioregionalization_metrics = NULL,
data_type = "abundance",
cluster_on = "site",
comat = vegemat,
similarity = NULL, # Name of similarity column
index = NULL,
verbose = FALSE)
wsb## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: occurrence
## - Metric data type: abundance
##
## Computed metrics:
## - Per-cluster co-occurrence metrics (abundance): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
##
## Data preview:
## $species_bioregions (11091 rows x 11 cols):
## Species Bioregion w_sb w_s w_b Specificity_abund NSpecificity_abund
## 10001 1 85 6255 1889243 0.014 0.007
## 10001 2 3037 6255 1081424 0.486 0.568
## 10001 3 3133 6255 1271474 0.501 0.425
## Fidelity_abund IndVal_abund NIndVal_abund Rho_abund
## 0.000 0.001 0.001 -6.806
## 0.003 0.314 0.368 4.731
## 0.002 0.315 0.267 3.256
## # ... with 11088 more rows
##
## Access data with:
## your_object$species_bioregionsIndVal (abundance)
\[IndVal_{sb} = A_{sb} \times \frac{n_{sb}}{n_b}\] Note that the fidelity based on occurrence is used here (De Cáceres & Legendre, 2009).
NIndVal (abundance)
\[\bar{IndVal}_{sb} = \bar{A}_{sb} \times \frac{n_{sb}}{n_b}\]
Note that the fidelity based on occurrence is used here (De Cáceres & Legendre, 2009).
Rho (abundance)
\[\rho_{sb} = \frac{\mu_{sb} - \mu_s}{\sqrt{\left(\frac{n - n_b}{n-1}\right) \left(\frac{{\sigma_s}^2}{n_b}\right)}}\] where
- \(\mu_{sb} = \frac{w_{sb}}{n_b}\) the average abundance of species \(s\) in bioregion \(b\) (as in NSpecificity and NIndVal)
- \(\mu_s = \frac{w_s}{n}\) the average abundance of species \(s\)
- \(\sigma_s\) the associated standard deviation.
7.2 Site metrics
For sites, two types of metrics can be computed, depending on whether the clustering is based on site or species:
- if the clustering is based on sites
(
cluster_on = "site"(or"both")), then richness and similarity-based metrics can be computed - if the clustering is based on species
(
cluster_on = "species"(or"both")), then we can also compute metrics that are typically applied at the species level, such as affinity, fidelity, IndVal and other similar metrics. The conceptual interpretation differs in this case.
7.2.1 Diversity & endemicity site metrics
When clusters are assigned to sites (bioregions), we can compute basic diversity metrics:
- Richness = number of species in the site
- Rich_Endemics = number of species in the site that are endemic to a single region (i.e., occur in only one bioregion)
- Prop_Endemics = proportion of endemic species, i.e. ratio between Rich_Endemics and Richness
sim_metrics <- site_species_metrics(bioregionalization = vege_nhclu,
bioregion_metrics = c("Richness", "Rich_Endemics",
"Prop_Endemics"),
bioregionalization_metrics = NULL,
data_type = "occurrence",
cluster_on = "site",
comat = vegemat,
similarity = vegesim,
index = "Simpson", # Name of similarity column
verbose = FALSE)
sim_metrics## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: occurrence
##
## Computed metrics:
## - Per-cluster richness & endemism metrics: Richness, Rich_Endemics, Prop_Endemics
##
## Data preview:
## $site_bioregions (2145 rows x 5 cols):
## Site Bioregion Richness Rich_Endemics Prop_Endemics
## 35 1 129 0 0.000
## 35 2 129 2 0.016
## 35 3 129 0 0.000
## # ... with 2142 more rows
##
## Access data with:
## your_object$site_bioregions7.2.2 Similarity-based site metrics
To compute similarity-based metrics for sites, we need to provide the
site similarity matrix (vegesim).
These metrics include the average similarity of each site to the
sites of
each bioregion (\(MeanSim\)) and the
associated standard deviation (\(SdSim\)).
When computing the average similarity, the focal site itself is not
included
in the calculation for its own bioregion.
sim_metrics <- site_species_metrics(bioregionalization = vege_nhclu,
bioregion_metrics = c("MeanSim", "SdSim"),
bioregionalization_metrics = NULL,
data_type = "occurrence",
cluster_on = "site",
comat = vegemat,
similarity = vegesim,
index = "Simpson", # Name of similarity column
verbose = FALSE)
sim_metrics## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: occurrence
##
## Computed metrics:
## - Per-cluster similarity-based metrics: MeanSim, SdSim
##
## Data preview:
## $site_bioregions (2145 rows x 4 cols):
## Site Bioregion MeanSim SdSim
## 35 1 0.055 0.033
## 35 2 0.554 0.340
## 35 3 0.251 0.195
## # ... with 2142 more rows
##
## Access data with:
## your_object$site_bioregionsMeanSim
Let \(g\) be a site and \(b\) a bioregion with sites \(g' \in b\), then:
\[MeanSim_{gb} = \frac{1}{n_b - \delta_{g \in b}} \sum_{g' \in b, g' \neq g} sim_{gg'}\] where \(sim_{gg'}\) is the similarity between sites \(g\) and \(g'\), \(n_b\) is the number of sites in bioregion \(b\), and \(\delta_{g \in b}\) is 1 if site \(g\) belongs to bioregion \(b\) (to exclude itself), 0 otherwise.
SdSim
The standard deviation of similarities of site \(g\) to bioregion \(b\) is:
\[SdSim_{gb} = \sqrt{\frac{1}{n_b - 1 - \delta_{g \in b}} \sum_{g' \in b, g' \neq g} \left( sim_{gg'} - MeanSim_{gb} \right)^2}\] where \(sim_{gg'}\) is the similarity between sites \(g\) and \(g'\), \(n_b\) is the number of sites in bioregion \(b\), and \(\delta_{g \in b}\) is 1 if site \(g\) belongs to bioregion \(b\) (to exclude itself), 0 otherwise.
7.2.3 Chorotype/Cluster-based site metrics
In the following example we compute only metrics for sites, on the
basis of species clusters (cluster_on = "species").
gc <- site_species_metrics(bioregionalization = vege_netclubip,
bioregion_metrics = c("Specificity", "NSpecificity",
"Fidelity",
"IndVal", "NIndVal",
"Rho",
"CoreTerms"),
bioregionalization_metrics = "P",
data_type = "both",
cluster_on = "species",
comat = vegemat,
similarity = NULL,
index = NULL,
verbose = FALSE)
gc## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: species
## - Clustering data type: abundance
## - Metric data type: occurrence and abundance
##
## Computed metrics:
## - Per-cluster co-occurrence metrics (occurrence): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
## - Per-cluster co-occurrence metrics (abundance): Specificity, NSpecificity, Fidelity, IndVal, NIndVal, Rho, CoreTerms
## - Bioregionalization co-occurrence metrics (occurrence): P
## - Bioregionalization co-occurrence metrics (abundance): P
##
## Data preview:
## $site_chorotypes (5720 rows x 20 cols):
## Site Chorotypes n_gc n_g n_c Specificity_occ NSpecificity_occ Fidelity_occ
## 35 1 1 129 2219 0.008 0.003 0.000
## 35 2 121 129 873 0.938 0.892 0.139
## 35 3 7 129 430 0.054 0.105 0.016
## IndVal_occ NIndVal_occ Rho_occ w_gc w_g w_c Specificity_abund
## 0.000 0.000 -13.981 1 423 2873216 0.002
## 0.130 0.124 19.103 411 423 1149003 0.972
## 0.001 0.002 -2.237 11 423 103264 0.026
## NSpecificity_abund Fidelity_abund IndVal_abund NIndVal_abund Rho_abund
## 0.001 0 0.000 0.000 -7.973
## 0.948 0 0.135 0.131 11.313
## 0.051 0 0.000 0.001 -1.840
## # ... with 5717 more rows
##
## $site_chorological (715 rows x 3 cols):
## Site P_occ P_abund
## 35 0.117 0.055
## 36 0.465 0.199
## 37 0.350 0.082
## # ... with 712 more rows
##
## Access data with:
## your_object$site_chorotypes
## your_object$site_chorological8. Metrics over the entire bioregionalization (i.e., over all clusters)
8.1 Site metrics
Based on \(MeanSim\), it is possible
to derive aggregated metrics that assess
how well a site fits within its assigned bioregion relative to
others.
For now, only the Silhouette index (Rousseeuw, 1987) is proposed.
Silhouette
The Silhouette index for a site \(g\) is defined as:
\[Silhouette_g = \frac{a_g - b_g}{\max(a_g, b_g)}\]
where:
-
\(a_g\) is the average similarity
of site \(g\) to all other sites in its
own bioregion,
- \(b_g\) is the average similarity of site \(g\) to all sites belonging to the nearest bioregion.
This index reflects how strongly a site is associated with its assigned bioregion relative to the most similar alternative bioregion, ranging from -1, when the site may be misassigned (i.e., more similar to another bioregion than its own), to 1, when the site is well matched to its own bioregion, and around 0 when the site lies near the boundary between bioregions.
sil_metrics <- site_species_metrics(bioregionalization = vege_nhclu,
bioregion_metrics = NULL,
bioregionalization_metrics = "Silhouette",
data_type = "occurrence",
cluster_on = "site",
comat = vegemat,
similarity = vegesim,
index = "Simpson", # Name of similarity column
verbose = FALSE)
sil_metrics## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: occurrence
##
## Computed metrics:
## - Bioregionalization similarity-based metrics: Silhouette
##
## Data preview:
## $site_bioregionalization (715 rows x 2 cols):
## Site Silhouette
## 35 0.548
## 36 0.308
## 37 0.441
## # ... with 712 more rows
##
## Access data with:
## your_object$site_bioregionalizationSite participation coefficient
We can compute the participation coefficient \(P_s\) of a species \(s\) to the bioregionalization as described in (Denelle et al., 2020), available in both its occurrence and abundance versions.
These metrics measure whether a site has species from a single region or from multiple regions - useful when investigating transition zones (Leroy et al., 2019). There are ranging from 0 to 1. Values close to 0 indicate that the site only has species from a single chorotype (i.e., not a transition zone), whereas values close to 1 indicate that the site has species evenely distributed across multiple chorotypes (i.e., likely a transition zone).
P (occurrence)
\[ P_s = 1 - \sum_{k=1}^K \left(\frac{n_{sk}}{n_s}\right)^2 \]
p_occ_site <- site_species_metrics(bioregionalization = vege_netclubip,
bioregion_metrics = NULL,
bioregionalization_metrics = "P",
data_type = "occurrence",
cluster_on = "species",
comat = vegemat,
similarity = NULL,
index = "Simpson", # Name of similarity column
verbose = FALSE)
p_occ_site## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: species
## - Clustering data type: abundance
## - Metric data type: occurrence
##
## Computed metrics:
## - Bioregionalization co-occurrence metrics (occurrence): P
##
## Data preview:
## $site_chorological (715 rows x 2 cols):
## Site P_occ
## 35 0.117
## 36 0.465
## 37 0.350
## # ... with 712 more rows
##
## Access data with:
## your_object$site_chorologicalP (abundance)
\[ P_s = 1 - \sum_{k=1}^K \left(\frac{w_{sk}}{w_s}\right)^2 \]
p_ab_site <- site_species_metrics(bioregionalization = vege_netclubip,
bioregion_metrics = NULL,
bioregionalization_metrics = "P",
data_type = "abundance",
cluster_on = "species",
comat = vegemat,
similarity = NULL,
index = "Simpson", # Name of similarity column
verbose = FALSE)
p_ab_site## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: species
## - Clustering data type: abundance
## - Metric data type: abundance
##
## Computed metrics:
## - Bioregionalization co-occurrence metrics (abundance): P
##
## Data preview:
## $site_chorological (715 rows x 2 cols):
## Site P_abund
## 35 0.055
## 36 0.199
## 37 0.082
## # ... with 712 more rows
##
## Access data with:
## your_object$site_chorological8.2 Species metrics
We can compute the participation coefficient \(P_s\) of a species \(s\) to the bioregionalization for species as well.
P (occurrence)
\[ P_s = 1 - \sum_{k=1}^K \left(\frac{n_{sk}}{n_s}\right)^2 \]
p_occ_sp <- site_species_metrics(bioregionalization = vege_netclubip,
bioregion_metrics = NULL,
bioregionalization_metrics = "P",
data_type = "occurrence",
cluster_on = "site",
comat = vegemat,
similarity = NULL,
index = "Simpson", # Name of similarity column
verbose = FALSE)
p_occ_sp## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: abundance
## - Metric data type: occurrence
##
## Computed metrics:
## - Bioregionalization co-occurrence metrics (occurrence): P
##
## Data preview:
## $species_bioregionalization (3697 rows x 2 cols):
## Species P_occ
## 10001 0.598
## 10002 0.188
## 10003 0.068
## # ... with 3694 more rows
##
## Access data with:
## your_object$species_bioregionalizationP (abundance)
\[ P_s = 1 - \sum_{k=1}^K \left(\frac{w_{sk}}{w_s}\right)^2 \]
p_ab_sp <- site_species_metrics(bioregionalization = vege_netclubip,
bioregion_metrics = NULL,
bioregionalization_metrics = "P",
data_type = "abundance",
cluster_on = "site",
comat = vegemat,
similarity = NULL,
index = "Simpson", # Name of similarity column
verbose = FALSE)
p_ab_sp## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: abundance
## - Metric data type: abundance
##
## Computed metrics:
## - Bioregionalization co-occurrence metrics (abundance): P
##
## Data preview:
## $species_bioregionalization (3697 rows x 2 cols):
## Species P_abund
## 10001 0.509
## 10002 0.090
## 10003 0.022
## # ... with 3694 more rows
##
## Access data with:
## your_object$species_bioregionalizationThese metrics measure how evenly a species is distributed among bioregions. There are ranging from 0 to 1. Values close to 0 indicate that the species is largely restricted to a single bioregion, while values close to 1 indicate that the species is evenly distributed across multiple bioregions.
Calculations on both occurrence & abundance at the same time:
ps <- site_species_metrics(bioregionalization = vege_nhclu,
bioregion_metrics = NULL,
bioregionalization_metrics = "P",
data_type = "both",
cluster_on = "site",
comat = vegemat,
similarity = NULL,
index = NULL,
verbose = FALSE)
ps## Site and species metrics
## ========================
##
## Settings:
## - Number of partitions: 1
## - Clusters based on: site
## - Clustering data type: occurrence
## - Metric data type: occurrence and abundance
##
## Computed metrics:
## - Bioregionalization co-occurrence metrics (occurrence): P
## - Bioregionalization co-occurrence metrics (abundance): P
##
## Data preview:
## $species_bioregionalization (3697 rows x 3 cols):
## Species P_occ P_abund
## 10001 0.581 0.513
## 10002 0.460 0.413
## 10003 0.089 0.022
## # ... with 3694 more rows
##
## Access data with:
## your_object$species_bioregionalization9. Bioregion metrics & spatial coherence
At the bioregion level, we can compute the number of sites and the
total
species richness across those sites. We can also derived the number
and
proportion of endemic species, defined (as above) as species occurring
exclusively
within a single bioregion (i.e., not found in any other bioregion).
bioregion_summary <- bioregion_metrics(vege_nhclu,
comat = vegemat)
bioregion_summary## Bioregion NbSites Richness Rich_Endemics Prop_Endemics
## 1 1 358 2821 407 0.14427508
## 2 2 150 2688 133 0.04947917
## 3 3 207 3090 58 0.01877023We also use the metric of spatial coherence following (Divíšek et al., 2016).
Spatial coherence quantifies how contiguous the sites of a bioregion
are.
Two variants are computed:
SC_size: the fraction of the bioregion’s sites contained in its largest
contiguous patch, calculated based on site counts rather than area.SC_area: the fraction of the bioregion’s total area contained in its
largest spatially contiguous patch, calculated from polygon geometries.
Both metrics range from 0 to 1, with 1 indicating a fully contiguous bioregion. Here is an example with the vegetation dataset.
data(vegesf)
bioregion_metrics(vege_nhclu,
comat = vegemat,
map = vegesf) ## Bioregion NbSites Richness Rich_Endemics Prop_Endemics SC_Size SC_Area
## 1 1 358 2821 407 0.14427508 0.9972067 0.9972067
## 2 2 150 2688 133 0.04947917 0.5400000 0.5400000
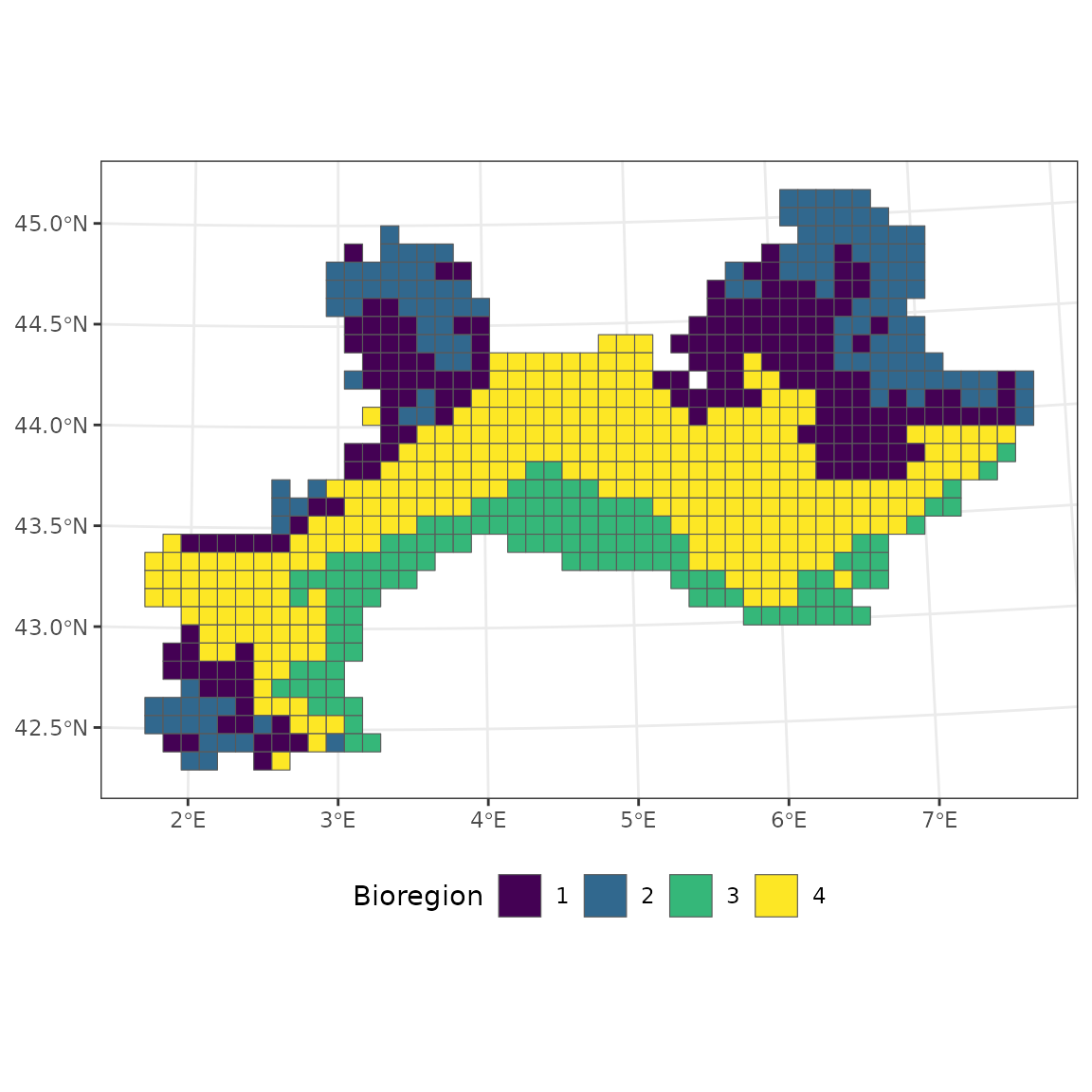
## 3 3 207 3090 58 0.01877023 0.5555556 0.5555556The bioregion 1 is almost constituted of one homogeneous block, which is why the spatial coherence is very close to 1.
map_bioregions(vege_nhclu,
map = vegesf)