Tutorial for bioregion
Maxime Lenormand, Boris Leroy and Pierre Denelle
2026-07-21
Source:vignettes/bioregion.Rmd
bioregion.Rmd0. Brief introduction
This tutorial aims at describing the different features of the R
package bioregion. The main purpose of the
bioregion‘s package is to propose a transparent
methodological framework to compare bioregionalization methods. Below is
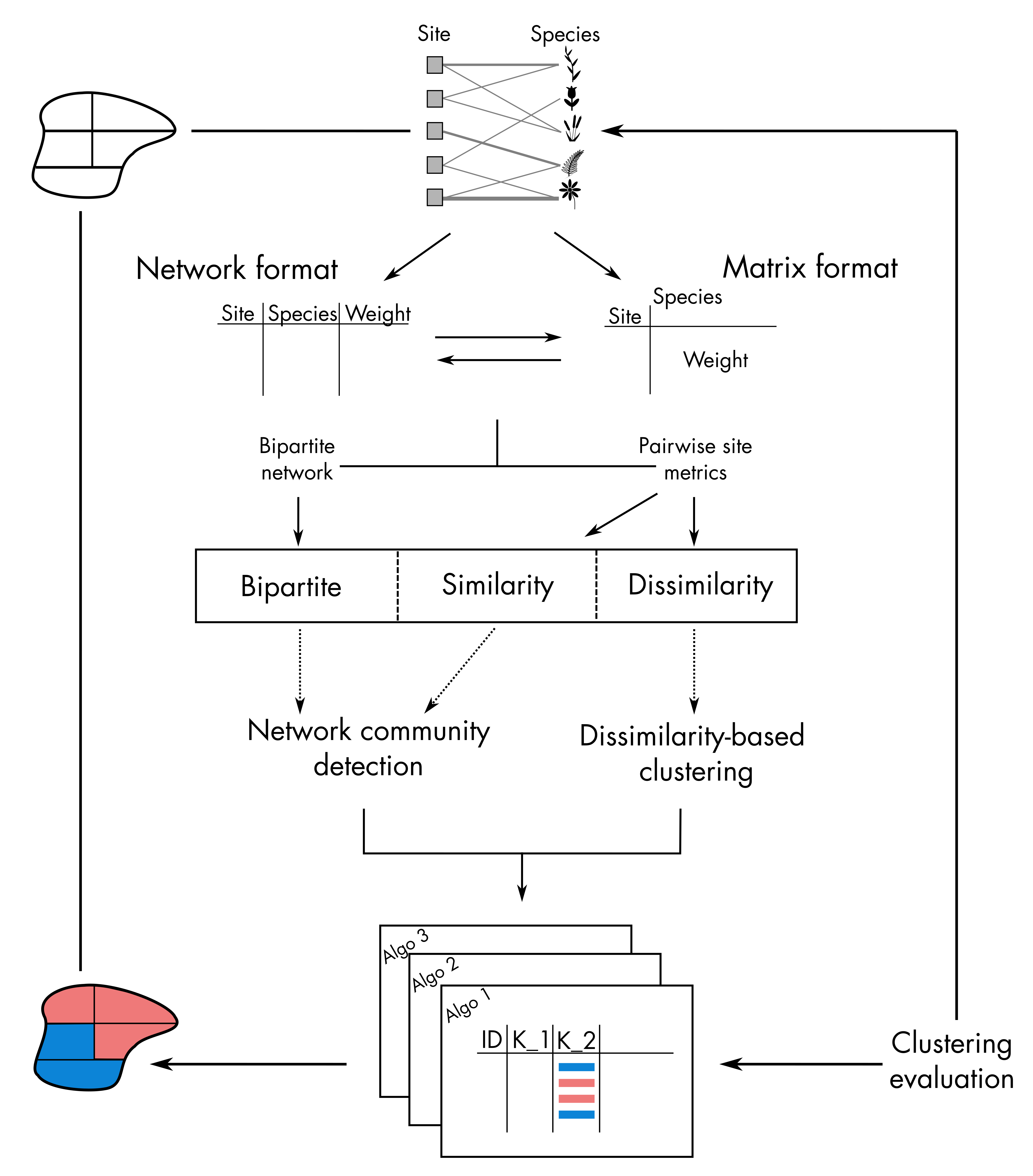
the typical flow chart of bioregions’ identification based on a
site-species bipartite network or co-occurrence matrix with
bioregion (Figure 1). This workflow can be divided into
four main steps:
- Preprocess the data (matrix or network formats)
- Compute similarity/dissimilarity metrics between sites based on species composition
- Run the different algorithms to identify different set of bioregions
- Evaluate and visualize the results
Please see this tutorial page for notes on the terminology used in the package.
1. Install binary files
Some functions or at least part of them (listed below) require binary files to run.
- netclu_infomap
- netclu_louvain (Cpp version)
- netclu_oslom
Please see this tutorial page to get instructions regarding the installation of the binary files.
2. Matrix or network formats
The bioregion’s package takes as input site-species
information stored in a bipartite network or a co-occurrence matrix.
Relying on the function mat_to_net
and net_to_mat
, it handles both the matrix and network formats throughout the
workflow.
Please see this tutorial page to better understand how these two functions work.
3. Pairwise similarity/dissimilarity metrics
The functions similarity
and dissimilarity
compute respectively pairwise similarity and dissimilarity metrics based
on a (site-species) co-occurrence matrix. The resulting
data.frame is stored in a bioregion.pairwise
object containing all requested metrics between each pair of sites.
The functions dissimilarity_to_similarity and similarity_to_dissimilarity can be used to transform a similarity object into a dissimilarity object and vice versa.
The function as_bioregion_pairwise
allows to convert a (dis)similarity matrix or a
list of such matrices into a
bioregion.pairwise object compatible with the
bioregion package. The input can come from base R,
dist objects, or outputs from other packages.
Please see this tutorial page to better understand how these functions work.
4. Bioregionalization algorithms
The bioregion R package gathers several methods allowing
to group sites and species into similar entities called bioregions. All
these methods can lead to several partitions of sites and species,
i.e. to different bioregionalizations.
Bioregionalization methods
can be based on hierarchical clustering algorithms, non-hierarchical
clustering algorithms or network algorithms.
The functions in the
package are related to each of these three families and produce output
that have a specific class, namely the bioregion.clusters
class.
4.1 Hierarchical clustering
The functions relying on hierarchical clustering start with the
prefix hclu_. With these algorithms, the bioregions are
placed into a dendrogram that ranges from two extremes: all sites belong
to the same bioregion (top of the tree) or all sites belong to a
different bioregion (bottom of the tree).
Please see this tutorial page for more details.
4.2 Non-hierarchical clustering
The functions relying on hierarchical clustering start with the
prefix nhclu_. For most of these algorithms, the user needs
to predefine the number of clusters, although this number can be
determined by estimating the optimal bioregionalization.
Please see this tutorial page for more details.
4.3 Network clustering
The functions relying on network clustering start with the prefix
netclu_. Site-species matrices can be seen as (bipartite)
networks where the nodes are either the sites or the species and the
links between them are the occurrences of species within sites.
With networks, modularity algorithms can be applied, leading to
bioregionalization.
The following tutorial page details more each clustering functions relying on a network algorithm.
4.4 Microbenchmark
The different bioregionalization methods listed in the package rely on more or less computationally intensive algorithms.
The following page estimates the time required to run each method on data sets of different sizes.
5.1 Visualization
If sites have geographic coordinates, then each bioregionalization
can be visualized with the function map_bioregions().
This tutorial page details different ways to plot your bioregionalization.
5.2 Summary metrics
In this section, we compute summary statistics at different scales, either at the bioregion or at the site or species level. Related functions are detailed in this page.
5.3 Compare bioregionalizations
In this section, we look at how sites are assigned to bioregions within a single bioregionalization and also compare this assignment across different bioregionalizations. The following page illustrates this.